
A couple of days ago one of the guys in the lab alerted my attention to a most interesting paper published in Organic Letters by Baldwin and co-workers from Oxford University. It's concerned with the postulated synthesis of a truncated analogue of
Spiculoic Acid A by
Mehta and
Kundu. But before I get into this latest paper from Baldwin and his mates let's go back and see how it all started.
It all took off in 2005 when
Mehta and
Kundu from the Indian Institute of Science in Bangalore published the following paper:
-
Toward a Total Synthesis of the Novel Polyketide Natural Product Spiculoic Acid A
Goverdhan Mehta and Uday Kumar Kundupp, Org. Lett., 2005, pp. 5569 - 5572
-
Now when I read this paper it actually came across as a nice piece of synthetic work. Unusually, these guys blatantly admit that their synthetic strategy towards the natural product failed. So what they do instead is provide a proof of concept by synthesising an analogue of the natural product using some Diels-Alder chemistry. Okay so that's all fine and dandy. At this point I'd like to say that I am very glad that I didn't referee this paper because things are about to get very hairy. Moving swiftly on to 2006 where Baldwin and co-workers publish the total synthesis of the enantiomer of Spiculonic Acid A in Chemical Communications:
-
Biomimetic synthesis of marine sponge metabolite spiculoic acid A and establishment of the absolute configuration of the natural product
James E. D.
Kirkham, Victor Lee and Jack E. Baldwin, Chem.
Commun., 2006, p. 2863
DOI:
10.1039/b607035c-
A very nice piece of synthetic work and a well written paper too. These dudes at Oxford really know what they are doing. So at this point I guess that Baldwin and his mates realised that there were some discrepancies between their data and those of Mehta and Kundu. Hence, they decided to sit down and dissect Mehta and Kundu's paper to figure out what was going on. The result of this little exercise was published in Organic Letters recently:
-
Stereochemical Reassignment of Mehta and Kundu's Spiculoic Acid A Analogue
Kirkham J. E. D., Lee, V. and Baldwin, J. E., Org.
Lett., 2006, ASAP Article
DOI:
10.1021/ol062361a -
Now this paper is really worth a read. We are talking major bitch slapping here. To me the most unbelievable mistake is the incorrect
stereochemical assignment of an
epoxide obtained by a
Sharpless asymmetric
epoxidation.
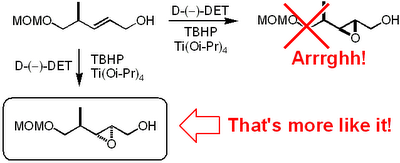
It appears that these Indian dudes haven't been able to use the mnemonic model published by
Sharpless to predict the
stereochemical outcome. This is what
Mehta and
Kundu write in their paper regarding the
epoxidation:
-
Sharpless epoxidation of allylic alcohol 19 in the presence of the D-tartaric acid diethyl ester was stereoselective (9:1) and afforded the epoxide 20 in a predictable manner with ample precedence.
-
And that's only the beginning. Their NOE interpretations are all over the place and it seems that they can't decide on the final stereochemistry of their Spiculonic Acid A when you compare the structures in the supplementary material with those given in the paper. Here's another brilliant quote from Mehta and Kundu's paper regarding their NOE interpretations (notice the language. One of these guys must have spent some time in the US and bought himself a dictionary):
-
The stereostructure of 9 was delineated on the basis of incisive analyses of its spectral characteristics, particularly the COSY and nOe data.
-
Anyway, hats off to Baldwin and co-workers for spotting all the mistakes and submitting the paper and to Organic Letters for accepting it. It is quite remarkable to think that these guys from Oxford have managed to publish in Organic Letters without conducting a single experiment. I highly recommend reading these three papers in chronological order. D!



 The Cambridge NMR service
The Cambridge NMR service  D!
D! A very important detail is that the template is a simple, achiral unit. The sole purpose of the template is to coordinate the Lewis acid well, exert rotamer control and as a consequence give good facial selective for the incoming nucleophile. Now as I mentioned before this principal works very well for many reactions. The Lewis acid is used in sub-stoichiometric quantities (generally 10-20 mol%). The radical stuff that I outlined above is okay cool but I personally like his stuff on pericyclic reactions better. Back in 2001 he published a very interesting paper in JACS (DOI:
A very important detail is that the template is a simple, achiral unit. The sole purpose of the template is to coordinate the Lewis acid well, exert rotamer control and as a consequence give good facial selective for the incoming nucleophile. Now as I mentioned before this principal works very well for many reactions. The Lewis acid is used in sub-stoichiometric quantities (generally 10-20 mol%). The radical stuff that I outlined above is okay cool but I personally like his stuff on pericyclic reactions better. Back in 2001 he published a very interesting paper in JACS (DOI: 

 This time there's also regioselectivity issues. However, they solve this and all other associated problems elegantly producing the desired dihydropyrazoles in excellent yields and ee's.
This time there's also regioselectivity issues. However, they solve this and all other associated problems elegantly producing the desired dihydropyrazoles in excellent yields and ee's.
 As Banwell pointed out the example with styrene is unbelievable and the ee's are through the roof! Moreover, the concept isn't limited to monosubstituted systems and more than 250 metabolites of this kind are known by now. So a couple of things immediately spring to mind. What scale can you do this sort of thing on and how do you get the other enantiomer of your product if that is what you are after. Well Banwell was on top of things and addressed these matters during his talk. Firstly, this stuff can be done on big scale. In the case of bromo- and chlorobenzene they obtain 35 grams of stuff per litre of fermentation broth which is pretty damn impressive. If you want the other enantiomer things are a bit trickier. However, Allen et al. have developed a method where the enantioselectivity is switched by introducing an iodine substituent that can be removed after the dihydroxylation:
As Banwell pointed out the example with styrene is unbelievable and the ee's are through the roof! Moreover, the concept isn't limited to monosubstituted systems and more than 250 metabolites of this kind are known by now. So a couple of things immediately spring to mind. What scale can you do this sort of thing on and how do you get the other enantiomer of your product if that is what you are after. Well Banwell was on top of things and addressed these matters during his talk. Firstly, this stuff can be done on big scale. In the case of bromo- and chlorobenzene they obtain 35 grams of stuff per litre of fermentation broth which is pretty damn impressive. If you want the other enantiomer things are a bit trickier. However, Allen et al. have developed a method where the enantioselectivity is switched by introducing an iodine substituent that can be removed after the dihydroxylation: A nice and simple solution to a complex problem that was published in Chemical Communications in 1995 (DOI:
A nice and simple solution to a complex problem that was published in Chemical Communications in 1995 (DOI: 
 However, the stuff in the JACS paper is different. Firstly, it only works if the carboxylic acid is adjacent to the heteroatom. Secondly, palladium adds adjacent to the carboxylic acid via an electrophilic palladation followed by palladium migration and concomitant decarboxylation. Finally the generated palladium species undergoes reductive elimination to form the desired product like this:
However, the stuff in the JACS paper is different. Firstly, it only works if the carboxylic acid is adjacent to the heteroatom. Secondly, palladium adds adjacent to the carboxylic acid via an electrophilic palladation followed by palladium migration and concomitant decarboxylation. Finally the generated palladium species undergoes reductive elimination to form the desired product like this:  In other words quite a different mechanism that doesn't involve a transmetallation step.
In other words quite a different mechanism that doesn't involve a transmetallation step.



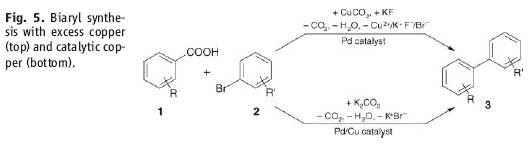






 I've shown the reaction for an alkyne starting material but it also works for alkenes to give alcohols rather than ketones.
I've shown the reaction for an alkyne starting material but it also works for alkenes to give alcohols rather than ketones. Now these Scandos did a thorough job and wrote a fairly detailed experimental procedure:
Now these Scandos did a thorough job and wrote a fairly detailed experimental procedure:
 3) Removing "part of the acetic acid" should be changed to: "remove most of the acetic acid". Otherwise, you'll end up doing nothing but adding sodium bicarbonate and filtering bucket loads of sodium acetate off for an entire day (just like I did!).
3) Removing "part of the acetic acid" should be changed to: "remove most of the acetic acid". Otherwise, you'll end up doing nothing but adding sodium bicarbonate and filtering bucket loads of sodium acetate off for an entire day (just like I did!).  Now you have to admit that this oxidation uses the coolest reagents ever just judged by their abbreviations.
Now you have to admit that this oxidation uses the coolest reagents ever just judged by their abbreviations. 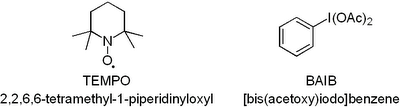 Both TEMPO and
Both TEMPO and  If you plan to hold on to it for a while it has to go in the freezer otherwise it goes off fast. Have fun, D!
If you plan to hold on to it for a while it has to go in the freezer otherwise it goes off fast. Have fun, D!

 Nice innit? So how much does it cost to get a kilo of TBDMS-Cl sent to the middle of nowhere? Well the actual product only sets you back 353 AU$ which is pretty damn cheap. However, the freight expenses are a bit out of control and amount to 308 AU$ . So the total price is 661 AU$ meaning it's ~6 times cheaper than to buy it from Aldrich.
Nice innit? So how much does it cost to get a kilo of TBDMS-Cl sent to the middle of nowhere? Well the actual product only sets you back 353 AU$ which is pretty damn cheap. However, the freight expenses are a bit out of control and amount to 308 AU$ . So the total price is 661 AU$ meaning it's ~6 times cheaper than to buy it from Aldrich. 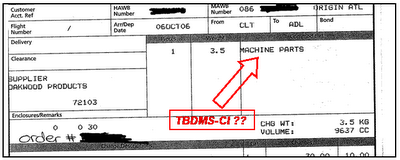 Hmmm very interesting. I guess it makes it a lot easier to get it through customs if you very conveniently forget to mention that it's 1 kilo of a flammable and corrosive chemical substance. Moreover, I noticed that it hasn't been synthesised in the US as I naively assumed.
Hmmm very interesting. I guess it makes it a lot easier to get it through customs if you very conveniently forget to mention that it's 1 kilo of a flammable and corrosive chemical substance. Moreover, I noticed that it hasn't been synthesised in the US as I naively assumed.


 orcid.org/0000-0003-3926-7047
orcid.org/0000-0003-3926-7047


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}