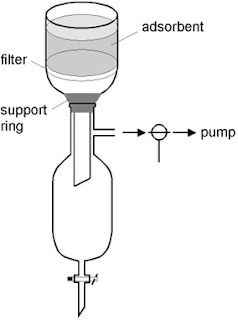
I get a lot of questions about Dry Column Vacuum Chromatography (DCVC) so I believe it is time for another post on the topic. DCVC is really taking off and the paper now has >70 journal citations.
Firstly, let me reiterate that like most things in life DCVC is best learnt by doing. Some of the questions I get are very detailed and specific and I can't provide clear cut answers. With experience you'll have an idea what to do and you'll improvise along the way and get it right.
-
Q&A session with the true believers:
-
(1) ...did you know that your paper had the"honor" of being "html-ed" by Rhodium.... - I didn't know that. I guess that everyone is purifying their illicit drugs in the garden shed using DCVC these days.
-
(2) ...searching for a good source for the silica gel, right now it is about 4-5 times more expensive than our flash gel source...
- That is a huge difference. You have a very sweet deal on flash silica. In my case it costs about 40 % more but considering how little you use compared to flash it works out as a big saving. If you find a cheap source of DCVC silica please let us know.
-
3) ...In the comments, you mentioned that you have done DCVC with 50-100 mg, and I was wondering what size of fritted funnel (frit diameter) you would use for that. Just calculating the size using the approximate amount of silica gel gives me something ridiculously small...
- The smallest sinter I use has a diameter of 1 cm. I have run 20 mg columns with 5 ml fractions on this column with no problems. Smaller than that would be impractical.
-
(4) ...Also, just to help me chose the size [Column] if I'd get one or two funnels made, what diameter(s) would you use for say 500 mg to 5 g?...
- I currently have the following four columns: 1, 4, 6 and 8 cm. My favourite (that I use 80% of the time) is 4 cm. It's a good size to work with and it's good for 20 ml fractions. I'll do anything from 100 mg and up to 5 grams or more (depending on the separation) on that column. For your requirements I'd say get a 4 and 6 cm column.
-
(5) ...frit porosities - the notation 1 - 4 is what is used in Europe, here in the US they have C, M and F, which actually don't directly correspond to the 1-4 sizes. "3" is a size where there is no direct equivalent, which is too bad because it seems ideal also to have in a flash column. The actual numbers are below, and I was a bit surprised because it seems that the P3 should clog up with time when using the Merck 15111 silica gel with its size range of 15-40 micron.
American standards - (Kimble & Corning, ASTM) nominal pore size, in microns, Medium 10-15 µm, Fine 4-5.5 µm
European standards - (Robu & Schott, ISO 4793) nominal pore size, in microns, P3 (P40) 16-40 µm, P4 (P16) 10-16 µm
- I had no idea about all this. Why on earth can't we just standardise these things. Anyway, thank you very much. I have always wondered exactly what the pore sizes represented.
-
(6) ...What is a least polar couple of spots with delta Rf 0.05 (often called "eight", "8") which you would separate via DCVC?...
- I only do DCVC. If it fails the next stop is prep. HPLC. Fortunately it still hasn't failed. As with all normal phase chromatography super non-polar compounds separation sucks. That said I have often columned stuff (not super non-polar) that appears to be one compound by TLC and managed to get two compounds of the column. Behold the power of slow step gradient elution. I have achieved truly mind boggling separations over only 20 fractions. The worst column ever occurred 1.5 years ago. I kept getting a fair bit of each diastereoisomer clean but it took 5 columns to get it fully separated. Still it was easily done in one day by using the same column 5 times and only collecting 30 fractions per column.
-
(7) Do you always start from heptane even for very polar mixtures?
- Yes! It gets the stuff off the Celite on to the silica and wets the column so that it runs well. Generally I do 4 x hexane, heptane or pet. ether first.
-
(8) ...can I use toluene/ethyl acetate, chloroform/ethyl acetate or chloroform/methanol mixtures successfully?...
- Yes! You can even use really low boiling solvents such as acetone, dichloromethane, ether etc. but due to evaporation it is easier to work with higher boiling solvents. Where I work now hexane and heptane has been replaced with 40-60 petroleum spirit which I use for my columns without too much difficulty. Remember to have the pump exhaust in the fume hood, especially with the low boiling solvents.
-
(9) ...Does it work substantially better [with Celite] than preadsorbtion on silica gel?...
-Yes! Celite is easier to handle and it doesn't affect resolution.
-
(10) ...Does this trick with Celite work even if a sample is only sparingly soluble in an eluent?...
- It doesn't matter what your eluent is. Dissolve your compound in something polar that is easy to get rid of, for example ethyl acetate or methanol. Add Celite, concentrate in vacuo and load it on the column. Ensure that you have removed all solvent prior to loading. A lot of say methanol in the Celite will compromise resolution.
-
I hope that helps. If you are new to this area please read the previous post and check out the following paper (a copy can be supplied upon request):
-
Dry Column Vacuum Chromatography, D.S. Pedersen and C. Rosenbohm, Synthesis, 2001, pp. 2431-2434.
-
Let me know if you have more comment, suggestions or questions. D!
 Not surprisingly, most of Curly Arrows readers are early career chemists, predominantly PhD students and Post Docs. So although subjects such as publications, H index, impact factors etc. have been beaten to death elsewhere I thought I'd do a brief post on the topic here because it is very important for your career prospects that you start thinking of these things early on. When in the past I have been presented with a pile of job applications the first things I (and others) look at are:
Not surprisingly, most of Curly Arrows readers are early career chemists, predominantly PhD students and Post Docs. So although subjects such as publications, H index, impact factors etc. have been beaten to death elsewhere I thought I'd do a brief post on the topic here because it is very important for your career prospects that you start thinking of these things early on. When in the past I have been presented with a pile of job applications the first things I (and others) look at are:









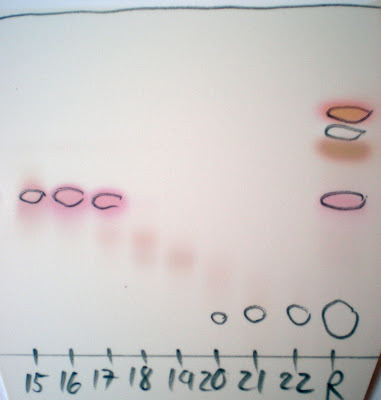 After treatment with a heat gun Ninhydrin Stain tends to give brightly coloured pink to purple spots as shown above. The spots can fade rather fast so record the result immediately. In the past I have been using a Ninhydrin Stain that didn't contain water, however adding a bit of water seems to improve the result a fair bit. D!
After treatment with a heat gun Ninhydrin Stain tends to give brightly coloured pink to purple spots as shown above. The spots can fade rather fast so record the result immediately. In the past I have been using a Ninhydrin Stain that didn't contain water, however adding a bit of water seems to improve the result a fair bit. D!
 So when Steven Ley comes up with a method that involves scooping solid magnesium nitride into a flask with your ester and some methanol, heating it to 80 oC for 24 hr, work up, filter, done! then that is really exciting good news to the synthetic organic chemist.
So when Steven Ley comes up with a method that involves scooping solid magnesium nitride into a flask with your ester and some methanol, heating it to 80 oC for 24 hr, work up, filter, done! then that is really exciting good news to the synthetic organic chemist.-the-anatomy-lecture-of-dr-nicolaes-tulp.jpg) Slides:
Slides:
 As always someone else got the idea first and where I work now melting point determinations are fully automated using an
As always someone else got the idea first and where I work now melting point determinations are fully automated using an 










 Pretty sweet and good quality chocolates too. Apparently, Fluka is a Swiss based company and once a year (around Christmas I believe) they give their customers a truck load of these containers.
Pretty sweet and good quality chocolates too. Apparently, Fluka is a Swiss based company and once a year (around Christmas I believe) they give their customers a truck load of these containers. One of the guys in the lab immediately grabbed the phone and gave Fluka Australia a call to hear if they stocked this item too. Unfortunately, this appears to be a Switzerland only bonus. The product also isn't listed in their catalogues.
One of the guys in the lab immediately grabbed the phone and gave Fluka Australia a call to hear if they stocked this item too. Unfortunately, this appears to be a Switzerland only bonus. The product also isn't listed in their catalogues.









 orcid.org/0000-0003-3926-7047
orcid.org/0000-0003-3926-7047


{kind=link}
{kind=link}
{kind=link}