A while back Chris published a post on a Science paper entitled:
-
Synthesis of Biaryls via Catalytic Decarboxylative Coupling (DOI: 10.1126/science.1128684).
-
A very interesting piece of work. However, then one our readers known as aa posted the following comment:
-
"Not sure if this is referenced in the Science paper (should be), but similar work was recently reported by a group at Boehringer Ingelheim in JACS. Check out JACS, 2006, 128, 11350-11351. They use palladium catalysis, and heteroaryl carboxylic acids, but the principle is identical."
-
-
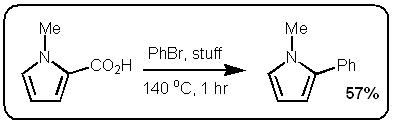
I finally did something about it and read the JACS paper (DOI:10.1021/ja063511f). Now first of all the German dudes who published their stuff in Science would have been hard pressed to cite the Boehringer Ingelheim group since their manuscript was submitted a month before the Boehringer people got round to it. The JACS paper is however very interesting. In fact, I find it even more exciting than the Science paper because these guys are making some pretty nice 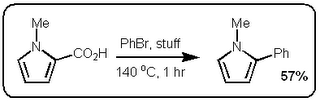 heteroaromatic systems that would make any pharma medchem person wet his pants. Moreover, they are doing it with excellent selectivity and in good yield. Most of the stuff in the paper is done under microwave conditions but they do one old school thermal example that works okay (See Scheme above).
heteroaromatic systems that would make any pharma medchem person wet his pants. Moreover, they are doing it with excellent selectivity and in good yield. Most of the stuff in the paper is done under microwave conditions but they do one old school thermal example that works okay (See Scheme above).
 heteroaromatic systems that would make any pharma medchem person wet his pants. Moreover, they are doing it with excellent selectivity and in good yield. Most of the stuff in the paper is done under microwave conditions but they do one old school thermal example that works okay (See Scheme above).
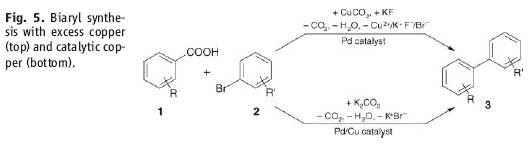
heteroaromatic systems that would make any pharma medchem person wet his pants. Moreover, they are doing it with excellent selectivity and in good yield. Most of the stuff in the paper is done under microwave conditions but they do one old school thermal example that works okay (See Scheme above).So at first glance these two papers seem very similar. However the postulated mechanisms are quite different. Firstly, the stuff in the Science paper starts of with a decarboxylation / copper insertion followed by a transmetallation and so forth. So in other words all the action is happening where the new bond is being made, like this:  However, the stuff in the JACS paper is different. Firstly, it only works if the carboxylic acid is adjacent to the heteroatom. Secondly, palladium adds adjacent to the carboxylic acid via an electrophilic palladation followed by palladium migration and concomitant decarboxylation. Finally the generated palladium species undergoes reductive elimination to form the desired product like this:
However, the stuff in the JACS paper is different. Firstly, it only works if the carboxylic acid is adjacent to the heteroatom. Secondly, palladium adds adjacent to the carboxylic acid via an electrophilic palladation followed by palladium migration and concomitant decarboxylation. Finally the generated palladium species undergoes reductive elimination to form the desired product like this:
However, the stuff in the JACS paper is different. Firstly, it only works if the carboxylic acid is adjacent to the heteroatom. Secondly, palladium adds adjacent to the carboxylic acid via an electrophilic palladation followed by palladium migration and concomitant decarboxylation. Finally the generated palladium species undergoes reductive elimination to form the desired product like this:  In other words quite a different mechanism that doesn't involve a transmetallation step.
In other words quite a different mechanism that doesn't involve a transmetallation step.Anyway, thanks to aa for the tip. This has been most educational. D!










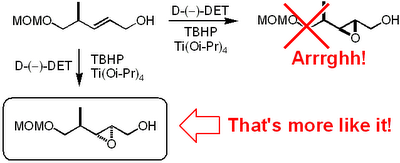 It appears that these Indian dudes haven't been able to use the mnemonic model published by
It appears that these Indian dudes haven't been able to use the mnemonic model published by  I've shown the reaction for an alkyne starting material but it also works for alkenes to give alcohols rather than ketones.
I've shown the reaction for an alkyne starting material but it also works for alkenes to give alcohols rather than ketones. Now these Scandos did a thorough job and wrote a fairly detailed experimental procedure:
Now these Scandos did a thorough job and wrote a fairly detailed experimental procedure:
 3) Removing "part of the acetic acid" should be changed to: "remove most of the acetic acid". Otherwise, you'll end up doing nothing but adding sodium bicarbonate and filtering bucket loads of sodium acetate off for an entire day (just like I did!).
3) Removing "part of the acetic acid" should be changed to: "remove most of the acetic acid". Otherwise, you'll end up doing nothing but adding sodium bicarbonate and filtering bucket loads of sodium acetate off for an entire day (just like I did!).  Now you have to admit that this oxidation uses the coolest reagents ever just judged by their abbreviations.
Now you have to admit that this oxidation uses the coolest reagents ever just judged by their abbreviations. 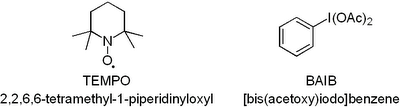 Both TEMPO and
Both TEMPO and  If you plan to hold on to it for a while it has to go in the freezer otherwise it goes off fast. Have fun, D!
If you plan to hold on to it for a while it has to go in the freezer otherwise it goes off fast. Have fun, D!


 orcid.org/0000-0003-3926-7047
orcid.org/0000-0003-3926-7047


{kind=link}
{kind=link}
{kind=link}
{kind=link}