For most academics, research can be a somewhat slow process. From the conception of an idea to actually getting started can take a significant amount of time. The topic of this post started as an idea based on Dror et al.'s publication back in 2011 that provided some strong in silico evidence for the presence of so-called metastable binding sites (MBS). Explained in very basic terms the hypothesis is that ligands do not simply arrive in their binding pockets randomly but follow a path of low affininty binding sites that guide them to their destination. The report by Dror et al. provided some very compelling in silico evidence for the existence of MBS and planted the idea with us of making bitopic ligands that would simultanously target the orthosteric binding site (OBS) and a predicted MBS using the same pharmacophore. In principle this could lead to ligands with improved receptor subtype selectivity, higher affinity and slower off rates. We described the idea in a perspective paper in J. Med. Chem. in 2017 and you can also get a very basic idea of the principle in the figure below.

I was lucky enough to secure some funding from the Lundbeck Foundation Natural Sciences for developing these types of ligands back in 2015. A great funding scheme by the Lundbeck Foundation that they sadly stopped some years ago. Anyway, with the funding we managed to make this work take off and published our first paper on bitopic ligands this year in J. Med. Chem. From our study it is not clear if we have the predicted bitopic binding mode but we have some good indications that things are indeed working as hoped for. Even better we have another paper coming up in 2020 were we have very strong evidence for a bitopic binding mode with a MBS so I look forward to sharing that. The ligands that we synthesised in our paper were beta-blockers and they all have a classic beta-amino alcohol motif that is synthesised from glycidol as outlined below.

At first this may seem as a simple synthesis with a logical outcome. You activate the epoxide (optically active glycidol) with a sulfonyl leaving group, do a nucleophilc substitution with a phenolate, followed by ring-opening of the epoxide with isopropylamine. However, this only works with no stereochemical leakage thanks to Professor Barry Sharpless. In fact, it is rather tricky to make activated glycidol ring open strictly via a SN2 mechanism (= no stereochemical leakage) with no competitive SN2' reaction (= racemisation). Sharpless and co-workers solved this problem by screening various leaving groups and found that the meta-nosyl group did the trick. To my great pleasure Professor Erland Stevens from Davidson College noticed our publication and decided to use it for educational purposes posting a video on YouTube that explains the glycidol ring-opening reaction in detail. Great to see that our science can be used for educational purposes. D!








 So when Steven Ley comes up with a method that involves scooping solid magnesium nitride into a flask with your ester and some methanol, heating it to 80 oC for 24 hr, work up, filter, done! then that is really exciting good news to the synthetic organic chemist.
So when Steven Ley comes up with a method that involves scooping solid magnesium nitride into a flask with your ester and some methanol, heating it to 80 oC for 24 hr, work up, filter, done! then that is really exciting good news to the synthetic organic chemist. The work is very throrough and makes up an 11 page JACS paper (not including any experimental). Many chemists would probably have split this work up in two papers. It's really nice to see these guys decided to stick the whole story in one paper. In brief these guys discover that they can make trisubstituted vinyl-cyclopropanes in high yield, diastereoselectivity and enantioselectivity. Moreover, they can make both enantiomers of cyclopropane selectively by switching from endo- to exo-sulfur ylides. This table from the paper illustrates how sweet this stuff is:
The work is very throrough and makes up an 11 page JACS paper (not including any experimental). Many chemists would probably have split this work up in two papers. It's really nice to see these guys decided to stick the whole story in one paper. In brief these guys discover that they can make trisubstituted vinyl-cyclopropanes in high yield, diastereoselectivity and enantioselectivity. Moreover, they can make both enantiomers of cyclopropane selectively by switching from endo- to exo-sulfur ylides. This table from the paper illustrates how sweet this stuff is: Only "problem" here is that they are using stoichiometric sulfur ylide. However, they address this by developing a catalytic ylide cyclopropanation. The yields are not as impressive and the ee's are down to 50-80%. Still pretty cool and I bet these guys are working hard to improve the catalytic system. Finally, they decide to pull off a short and high yielding formal total synthesis of a known cyclopropane amino acid.
Only "problem" here is that they are using stoichiometric sulfur ylide. However, they address this by developing a catalytic ylide cyclopropanation. The yields are not as impressive and the ee's are down to 50-80%. Still pretty cool and I bet these guys are working hard to improve the catalytic system. Finally, they decide to pull off a short and high yielding formal total synthesis of a known cyclopropane amino acid. Obviously, they are making both enantiomers as well as both enantiomers of a diastereoisomer. And here I'm messing around trying to improve my lousy dr's on the racemic synthesis of the same target. Crap! D!
Obviously, they are making both enantiomers as well as both enantiomers of a diastereoisomer. And here I'm messing around trying to improve my lousy dr's on the racemic synthesis of the same target. Crap! D!

 As it turns out the chemistry works really well for most systems using catalyst 1. However, some aldehydes require catalyst 2 to give a good result, eg. entries 3 and 4. The only compounds tested in this paper that failed completely were aldehydes that exist predominantly in a hemiacetal form, eg. 5-hydroxy-valeraldehyde. So there you have it. Maybe something you should consider giving a go next time it's alpha-methylenation time. D!
As it turns out the chemistry works really well for most systems using catalyst 1. However, some aldehydes require catalyst 2 to give a good result, eg. entries 3 and 4. The only compounds tested in this paper that failed completely were aldehydes that exist predominantly in a hemiacetal form, eg. 5-hydroxy-valeraldehyde. So there you have it. Maybe something you should consider giving a go next time it's alpha-methylenation time. D!


 Nice stuff innit and it gets better.
Nice stuff innit and it gets better.
 Fortunately, all the ingredients are reasonably affordable. Mixing it all up and adding the alcohol gives a biphasic reaction mixture that looks a bit like Schweppes Orange:
Fortunately, all the ingredients are reasonably affordable. Mixing it all up and adding the alcohol gives a biphasic reaction mixture that looks a bit like Schweppes Orange: However, unlike Delfourne et al. my final product wasn't clean after a simple work up. Succinimide was simply precipitating everywhere and hence some silica was required. In the end a filtration through a silica plug proved sufficient to give clean product on a reasonably large scale (18 grams) in excellent yield (97%). So despite the fact that a simple work up wasn't sufficient to clean the product up this is an easy to do reaction that I would recommend to anyone who's tired of stinky old Swern. D!
However, unlike Delfourne et al. my final product wasn't clean after a simple work up. Succinimide was simply precipitating everywhere and hence some silica was required. In the end a filtration through a silica plug proved sufficient to give clean product on a reasonably large scale (18 grams) in excellent yield (97%). So despite the fact that a simple work up wasn't sufficient to clean the product up this is an easy to do reaction that I would recommend to anyone who's tired of stinky old Swern. D!  Now the above reaction is obviously totally irresponsible and was only on for less than a minute so that I could take the picture. We are dealing with a fancy piece of glassware that has oxygen bubbling through it whilst two flood lamps are hammering photons away generating singlet oxygen and in the process heating the fume hood up big time. Hence, aluminium foil on the bottom of the hood is required to
Now the above reaction is obviously totally irresponsible and was only on for less than a minute so that I could take the picture. We are dealing with a fancy piece of glassware that has oxygen bubbling through it whilst two flood lamps are hammering photons away generating singlet oxygen and in the process heating the fume hood up big time. Hence, aluminium foil on the bottom of the hood is required to  Well at least I know it's beautiful behind all that plastic and aluminium foil. This particular day I was doing the following photolysis:
Well at least I know it's beautiful behind all that plastic and aluminium foil. This particular day I was doing the following photolysis: 
 We tend to use the bis-
We tend to use the bis- A very important detail is that the template is a simple, achiral unit. The sole purpose of the template is to coordinate the Lewis acid well, exert rotamer control and as a consequence give good facial selective for the incoming nucleophile. Now as I mentioned before this principal works very well for many reactions. The Lewis acid is used in sub-stoichiometric quantities (generally 10-20 mol%). The radical stuff that I outlined above is okay cool but I personally like his stuff on pericyclic reactions better. Back in 2001 he published a very interesting paper in JACS (DOI:
A very important detail is that the template is a simple, achiral unit. The sole purpose of the template is to coordinate the Lewis acid well, exert rotamer control and as a consequence give good facial selective for the incoming nucleophile. Now as I mentioned before this principal works very well for many reactions. The Lewis acid is used in sub-stoichiometric quantities (generally 10-20 mol%). The radical stuff that I outlined above is okay cool but I personally like his stuff on pericyclic reactions better. Back in 2001 he published a very interesting paper in JACS (DOI: 

 This time there's also regioselectivity issues. However, they solve this and all other associated problems elegantly producing the desired dihydropyrazoles in excellent yields and ee's.
This time there's also regioselectivity issues. However, they solve this and all other associated problems elegantly producing the desired dihydropyrazoles in excellent yields and ee's.
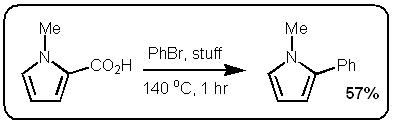
 However, the stuff in the JACS paper is different. Firstly, it only works if the carboxylic acid is adjacent to the heteroatom. Secondly, palladium adds adjacent to the carboxylic acid via an electrophilic palladation followed by palladium migration and concomitant decarboxylation. Finally the generated palladium species undergoes reductive elimination to form the desired product like this:
However, the stuff in the JACS paper is different. Firstly, it only works if the carboxylic acid is adjacent to the heteroatom. Secondly, palladium adds adjacent to the carboxylic acid via an electrophilic palladation followed by palladium migration and concomitant decarboxylation. Finally the generated palladium species undergoes reductive elimination to form the desired product like this:  In other words quite a different mechanism that doesn't involve a transmetallation step.
In other words quite a different mechanism that doesn't involve a transmetallation step. I've shown the reaction for an alkyne starting material but it also works for alkenes to give alcohols rather than ketones.
I've shown the reaction for an alkyne starting material but it also works for alkenes to give alcohols rather than ketones. Now these Scandos did a thorough job and wrote a fairly detailed experimental procedure:
Now these Scandos did a thorough job and wrote a fairly detailed experimental procedure:
 3) Removing "part of the acetic acid" should be changed to: "remove most of the acetic acid". Otherwise, you'll end up doing nothing but adding sodium bicarbonate and filtering bucket loads of sodium acetate off for an entire day (just like I did!).
3) Removing "part of the acetic acid" should be changed to: "remove most of the acetic acid". Otherwise, you'll end up doing nothing but adding sodium bicarbonate and filtering bucket loads of sodium acetate off for an entire day (just like I did!). 

 orcid.org/0000-0003-3926-7047
orcid.org/0000-0003-3926-7047


{kind=link}
{kind=link}