I have on several occasions been asked to make a DCVC video tutorial and quite liked the idea of doing so. Thus, I have started my acting career as you can see in the video below. I think the video will be a useful guide for first time DCVCers. For more info you should consult this and this blog post on DCVC. Many thanks to the University of Copenhagen's Communication Department, in particular Jacob Lejbach Sørensen, for investing some time in making this possible. D!
Showing posts with label chromatography. Show all posts
Showing posts with label chromatography. Show all posts
Monday, February 20, 2017
Tuesday, November 15, 2011
Reverse Phase Silica - Sort of!

I'm sure that all the synthetic organic chemists will agree that column chromatography is something we simply couldn't live without. At our Department we use it in all its forms: Automated flash chromatography, Chromatotron, Prep. HPLC (Chiral and RP), Prep. TLC, old skool manual flash and DCVC columns etc. However, in spite of all this we all too frequently end up with stuff that is a major pain to separate. Just last week a colleague of mine, lets call him Bernard, told me he had used deactivated silica for purifying his compound - the only thing that had worked for him! So now you are probably thinking deactivated silica??? Normal lab terminology translates this to treating your silica with some amine base prior to use but this is not what Bernard was talking about. He found this paper (Procedure in the Supp. Info.) where they derivatise silica gel with ethyltrichlorosilane. Cheap reagents and a simple method. I guess the "deactivated" silica ends up being something between regular and reverse phase silica. Bernard simply packed a column with this stuff and put on our automated columning system to get a great result eluting with normal phase solvent mixtures. Excellent news for me and my people. We often end up with very polar compounds that will not come off a regular column or elute in fractions 1-5 on a reverse phase column.
Does anyone have any experience with this type of silica or something similar? Please let us all know by posting a comment. D!
Friday, April 04, 2008
Dry Column Vacuum Chromatography (DCVC) - revisited
 I get a lot of questions about Dry Column Vacuum Chromatography (DCVC) so I believe it is time for another post on the topic. DCVC is really taking off and the paper now has >70 journal citations.
I get a lot of questions about Dry Column Vacuum Chromatography (DCVC) so I believe it is time for another post on the topic. DCVC is really taking off and the paper now has >70 journal citations.Firstly, let me reiterate that like most things in life DCVC is best learnt by doing. Some of the questions I get are very detailed and specific and I can't provide clear cut answers. With experience you'll have an idea what to do and you'll improvise along the way and get it right.
-
Q&A session with the true believers:
-
(1) ...did you know that your paper had the"honor" of being "html-ed" by Rhodium....
- I didn't know that. I guess that everyone is purifying their illicit drugs in the garden shed using DCVC these days.
-
(2) ...searching for a good source for the silica gel, right now it is about 4-5 times more expensive than our flash gel source...
- That is a huge difference. You have a very sweet deal on flash silica. In my case it costs about 40 % more but considering how little you use compared to flash it works out as a big saving. If you find a cheap source of DCVC silica please let us know.
-
3) ...In the comments, you mentioned that you have done DCVC with 50-100 mg, and I was wondering what size of fritted funnel (frit diameter) you would use for that. Just calculating the size using the approximate amount of silica gel gives me something ridiculously small...
- The smallest sinter I use has a diameter of 1 cm. I have run 20 mg columns with 5 ml fractions on this column with no problems. Smaller than that would be impractical.
-
(4) ...Also, just to help me chose the size [Column] if I'd get one or two funnels made, what diameter(s) would you use for say 500 mg to 5 g?...
- I currently have the following four columns: 1, 4, 6 and 8 cm. My favourite (that I use 80% of the time) is 4 cm. It's a good size to work with and it's good for 20 ml fractions. I'll do anything from 100 mg and up to 5 grams or more (depending on the separation) on that column. For your requirements I'd say get a 4 and 6 cm column.
-
(5) ...frit porosities - the notation 1 - 4 is what is used in Europe, here in the US they have C, M and F, which actually don't directly correspond to the 1-4 sizes. "3" is a size where there is no direct equivalent, which is too bad because it seems ideal also to have in a flash column. The actual numbers are below, and I was a bit surprised because it seems that the P3 should clog up with time when using the Merck 15111 silica gel with its size range of 15-40 micron.
American standards - (Kimble & Corning, ASTM) nominal pore size, in microns, Medium 10-15 µm, Fine 4-5.5 µm
European standards - (Robu & Schott, ISO 4793) nominal pore size, in microns, P3 (P40) 16-40 µm, P4 (P16) 10-16 µm
- I had no idea about all this. Why on earth can't we just standardise these things. Anyway, thank you very much. I have always wondered exactly what the pore sizes represented.
-
(6) ...What is a least polar couple of spots with delta Rf 0.05 (often called "eight", "8") which you would separate via DCVC?...
- I only do DCVC. If it fails the next stop is prep. HPLC. Fortunately it still hasn't failed. As with all normal phase chromatography super non-polar compounds separation sucks. That said I have often columned stuff (not super non-polar) that appears to be one compound by TLC and managed to get two compounds of the column. Behold the power of slow step gradient elution. I have achieved truly mind boggling separations over only 20 fractions. The worst column ever occurred 1.5 years ago. I kept getting a fair bit of each diastereoisomer clean but it took 5 columns to get it fully separated. Still it was easily done in one day by using the same column 5 times and only collecting 30 fractions per column.
-
(7) Do you always start from heptane even for very polar mixtures?
- Yes! It gets the stuff off the Celite on to the silica and wets the column so that it runs well. Generally I do 4 x hexane, heptane or pet. ether first.
-
(8) ...can I use toluene/ethyl acetate, chloroform/ethyl acetate or chloroform/methanol mixtures successfully?...
- Yes! You can even use really low boiling solvents such as acetone, dichloromethane, ether etc. but due to evaporation it is easier to work with higher boiling solvents. Where I work now hexane and heptane has been replaced with 40-60 petroleum spirit which I use for my columns without too much difficulty. Remember to have the pump exhaust in the fume hood, especially with the low boiling solvents.
-
(9) ...Does it work substantially better [with Celite] than preadsorbtion on silica gel?...
-Yes! Celite is easier to handle and it doesn't affect resolution.
-
(10) ...Does this trick with Celite work even if a sample is only sparingly soluble in an eluent?...
- It doesn't matter what your eluent is. Dissolve your compound in something polar that is easy to get rid of, for example ethyl acetate or methanol. Add Celite, concentrate in vacuo and load it on the column. Ensure that you have removed all solvent prior to loading. A lot of say methanol in the Celite will compromise resolution.
-
I hope that helps. If you are new to this area please read the previous post and check out the following paper (a copy can be supplied upon request):
-
Dry Column Vacuum Chromatography, D.S. Pedersen and C. Rosenbohm, Synthesis, 2001, pp. 2431-2434.
-
Let me know if you have more comment, suggestions or questions. D!
Thursday, February 07, 2008
Recycling Silica Gel
 Insert usual excuse for infrequent posting here: .....
Insert usual excuse for infrequent posting here: .....-
I recently received the following email regarding recycling of silica gel:
-
"In the past we used to regenerate SiGel with fuming nitric acid, much as you do here (refers to previous post - D!) .Place the coloured, used SiGel (which of course must be uncoated and completely dry) in a large beaker. Approximately a third full. Place it inthe fume cupboard and pour in the nitric acid so the SiGel is completely moistened. If necessary, stir it. After a possible initial fuming has stopped, heat it on a steam bath for 10 minutes - leave it overnight. Next day, fill up with water, stir and let it settle. Wash 3 more times. At this point, it should be colourless, if not, repeat with more nitric acid. Then wash with satd. sodium bicarbonate until neutral and filter on a Buchner funnel. Wash with water, methanol and acetone and let it suck completely dry. Finally, activate by heating it in an oven overnight at 100-120 C.This way, it is as good as new for most purposes."
-
This may be of use to some chemists working at institutions where money is very scarce. However, considering the amount of time and large volumes of solvent required to do this I think it can be classified as historically interesting but not practically useful to most of us.
-
Nevertheless thank you for the email. I have wondered exactly how you would go about recycling your silica gel.
-
Speaking of silica gel and techniques that are disappearing from the chemists hood check one of the most recent posts at In the Pipeline out. Derek seems to think that TLCs are on their way out as LC-MS is becoming more and more common. I believe that it will take several decades before Derek's predictions come true (especially in academia) but he does have some very good points. D!
Monday, March 05, 2007
A Column Apart
It's quiz time. Check out the flash column below. I may look like any other flash column but it is a truly unusual little fella. This is the first chromatography column of this kind I have ever put eyes on. I admired it for quite a while together with the other guys in the lab. We were all there for the packing and loading.  Amazing stuff...our presence really freaked the guy running the column out. I don't think he grasped the significance of what he was doing. Anyway, what do you think is so special about it? D!
Amazing stuff...our presence really freaked the guy running the column out. I don't think he grasped the significance of what he was doing. Anyway, what do you think is so special about it? D!
Amazing stuff...our presence really freaked the guy running the column out. I don't think he grasped the significance of what he was doing. Anyway, what do you think is so special about it? D!Thursday, November 16, 2006
Dry Column Vacuum Chromatography
Some years ago when I was silly enough to have a real job in the chemical industry we were working in a regular synthetic organic lab but had to scale some of our synthetic work up considerably. This makes everything a bit complicated and time consuming. Now you can get a long way by just buying massive RBF's and so forth and as long as you can recrystallise your products everyone is happy. However, when you have to run your 250 grams of stuff down a quick flash column you have a serious problem. So we were looking for some alternative to flash column chromatography that would allow us to do so. A good friend mine at the Australian Wine Research Institute of all places told me about something he called squat columns and gave me a paper from Aldrichimica Acta on the technique:
"Dry-column" Flash Chromatography, L.M. Harwood, Aldrichimica Acta, 1985, 18, p. 25
Now this paper isn't particularly detailed so we had to mess around with it for quite a long time before we got it right but it was worth it. We were columning really large quantities of stuff this way. I spoke to my former industry colleague about it the other day and he told me that they were doing 500 gram columns now collecting 1 and 2 liter fractions in conical flasks. Anyway, we thought that it made sense to publish a paper with more detailed instructions so that someone might actually try to use it successfully. Fortunately, the referees also liked the idea so we got this paper out on the topic: Dry Column Vacuum Chromatography, D.S. Pedersen and C. Rosenbohm, Synthesis, 2001, pp. 2431-2434
Dry Column Vacuum Chromatography, D.S. Pedersen and C. Rosenbohm, Synthesis, 2001, pp. 2431-2434
We even took the liberty of renaming the technique because we thought flash was a bit misleading. Basically Dry Column Vacuum Chromatography (DCVC) is an alternative to flash where the solvent is sucked through the column. The solvent is added one fractions at the time (making very precise gradient elution dead easy) and hence the column is sucked almost completely dry between fractions. For the full details read the paper. So what does it look like? Above there's a picture of a 20 gram column I ran the other day. Now this is a big column but it also works with small columns. My favourite size columns have a diameter of 4-6 cm. They are easy to work with and I would typically collect 20 ml fractions. If you want to give DCVC a go but don't want to ask the glass blower for a fancy piece of glassware at first you don't have to. Some of the bits and pieces you need for a simple set up are shown above. A nice and sturdy spatula is essential for good column packing. Quite a few chemist I know use water aspirators for vacuum rather than a fancy diaphragm pump and it seems to work for them so that's also an option. The trick to make this technique work is to get the right silica.
column but it also works with small columns. My favourite size columns have a diameter of 4-6 cm. They are easy to work with and I would typically collect 20 ml fractions. If you want to give DCVC a go but don't want to ask the glass blower for a fancy piece of glassware at first you don't have to. Some of the bits and pieces you need for a simple set up are shown above. A nice and sturdy spatula is essential for good column packing. Quite a few chemist I know use water aspirators for vacuum rather than a fancy diaphragm pump and it seems to work for them so that's also an option. The trick to make this technique work is to get the right silica.
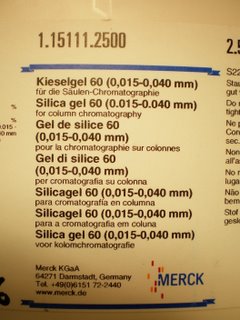 If you use the stuff to the right it will all work fine. However, if you use regular flash silica it will not work. Don't even try it's a waste of time. Regular flash silica is simply to coarse and you can't pack a proper column.
If you use the stuff to the right it will all work fine. However, if you use regular flash silica it will not work. Don't even try it's a waste of time. Regular flash silica is simply to coarse and you can't pack a proper column.
Cool features about DCVC are:
(1) Large scale columns are possible
(2) It's faster than flash. Mainly because you don't collect many fractions (usually 20-30)
(3) You don't use much silica (column is only ~5 cm high) or solvents
(4) Because the column gets sucked dry between fractions you can easily do other stuff whilst running it. Even leaving it for a few hours will not ruin the resolution.
My personal record is seven DCVC's in one day (didn't go home until 10 pm though). However, I wouldn't recommend doing more than four in a day. You start going insane after the fourth column. The technique works so well that I haven't run a flash column in five years and I have no intention of ever doing one again. The columns can normally be recycled. Just give them a good rinse. I usually do something like: MeOH, EtOAc and finally hexane. Seal it up nicely and stick in the fridge. They easily last for a week in the fridge if you seal them up properly. This is how I do it: An old suba seal on the stem a bit of para film on the top with a bit of tape and stick in the fridge in a beaker (see picture).
An old suba seal on the stem a bit of para film on the top with a bit of tape and stick in the fridge in a beaker (see picture).
One important detail that we have discovered since publishing the paper is that one should NOT concentrate the sample to be columned on silica. Instead, you should use Celite (or Kenite). Opposed to silica Celite is very non-polar and hence you can use quite a lot for loading you sample without compromising the resolution. This way you can ensure that you get as much of your compound as possible on to the actual column. If you stick a piece of filter paper on top of the silica column prior to loading the celite it makes it really easy for you to scrape the celite off after running the column so that you can use it once more. Also you should stick a piece of filter paper on top of the kenite so that you don't mess the surface up when you pour the solvent on. Okay I'd better stop here. You obviously need to read the paper before you try this method. Just send an email to curlyarrow@gmail.com to request a copy. D!
"Dry-column" Flash Chromatography, L.M. Harwood, Aldrichimica Acta, 1985, 18, p. 25
Now this paper isn't particularly detailed so we had to mess around with it for quite a long time before we got it right but it was worth it. We were columning really large quantities of stuff this way. I spoke to my former industry colleague about it the other day and he told me that they were doing 500 gram columns now collecting 1 and 2 liter fractions in conical flasks. Anyway, we thought that it made sense to publish a paper with more detailed instructions so that someone might actually try to use it successfully. Fortunately, the referees also liked the idea so we got this paper out on the topic:
 Dry Column Vacuum Chromatography, D.S. Pedersen and C. Rosenbohm, Synthesis, 2001, pp. 2431-2434
Dry Column Vacuum Chromatography, D.S. Pedersen and C. Rosenbohm, Synthesis, 2001, pp. 2431-2434We even took the liberty of renaming the technique because we thought flash was a bit misleading. Basically Dry Column Vacuum Chromatography (DCVC) is an alternative to flash where the solvent is sucked through the column. The solvent is added one fractions at the time (making very precise gradient elution dead easy) and hence the column is sucked almost completely dry between fractions. For the full details read the paper. So what does it look like? Above there's a picture of a 20 gram column I ran the other day. Now this is a big
 column but it also works with small columns. My favourite size columns have a diameter of 4-6 cm. They are easy to work with and I would typically collect 20 ml fractions. If you want to give DCVC a go but don't want to ask the glass blower for a fancy piece of glassware at first you don't have to. Some of the bits and pieces you need for a simple set up are shown above. A nice and sturdy spatula is essential for good column packing. Quite a few chemist I know use water aspirators for vacuum rather than a fancy diaphragm pump and it seems to work for them so that's also an option. The trick to make this technique work is to get the right silica.
column but it also works with small columns. My favourite size columns have a diameter of 4-6 cm. They are easy to work with and I would typically collect 20 ml fractions. If you want to give DCVC a go but don't want to ask the glass blower for a fancy piece of glassware at first you don't have to. Some of the bits and pieces you need for a simple set up are shown above. A nice and sturdy spatula is essential for good column packing. Quite a few chemist I know use water aspirators for vacuum rather than a fancy diaphragm pump and it seems to work for them so that's also an option. The trick to make this technique work is to get the right silica. If you use the stuff to the right it will all work fine. However, if you use regular flash silica it will not work. Don't even try it's a waste of time. Regular flash silica is simply to coarse and you can't pack a proper column.
If you use the stuff to the right it will all work fine. However, if you use regular flash silica it will not work. Don't even try it's a waste of time. Regular flash silica is simply to coarse and you can't pack a proper column.Cool features about DCVC are:
(1) Large scale columns are possible
(2) It's faster than flash. Mainly because you don't collect many fractions (usually 20-30)
(3) You don't use much silica (column is only ~5 cm high) or solvents
(4) Because the column gets sucked dry between fractions you can easily do other stuff whilst running it. Even leaving it for a few hours will not ruin the resolution.
My personal record is seven DCVC's in one day (didn't go home until 10 pm though). However, I wouldn't recommend doing more than four in a day. You start going insane after the fourth column. The technique works so well that I haven't run a flash column in five years and I have no intention of ever doing one again. The columns can normally be recycled. Just give them a good rinse. I usually do something like: MeOH, EtOAc and finally hexane. Seal it up nicely and stick in the fridge. They easily last for a week in the fridge if you seal them up properly. This is how I do it:
 An old suba seal on the stem a bit of para film on the top with a bit of tape and stick in the fridge in a beaker (see picture).
An old suba seal on the stem a bit of para film on the top with a bit of tape and stick in the fridge in a beaker (see picture).One important detail that we have discovered since publishing the paper is that one should NOT concentrate the sample to be columned on silica. Instead, you should use Celite (or Kenite). Opposed to silica Celite is very non-polar and hence you can use quite a lot for loading you sample without compromising the resolution. This way you can ensure that you get as much of your compound as possible on to the actual column. If you stick a piece of filter paper on top of the silica column prior to loading the celite it makes it really easy for you to scrape the celite off after running the column so that you can use it once more. Also you should stick a piece of filter paper on top of the kenite so that you don't mess the surface up when you pour the solvent on. Okay I'd better stop here. You obviously need to read the paper before you try this method. Just send an email to curlyarrow@gmail.com to request a copy. D!
Subscribe to:
Posts (Atom)


 orcid.org/0000-0003-3926-7047
orcid.org/0000-0003-3926-7047


{kind=link}