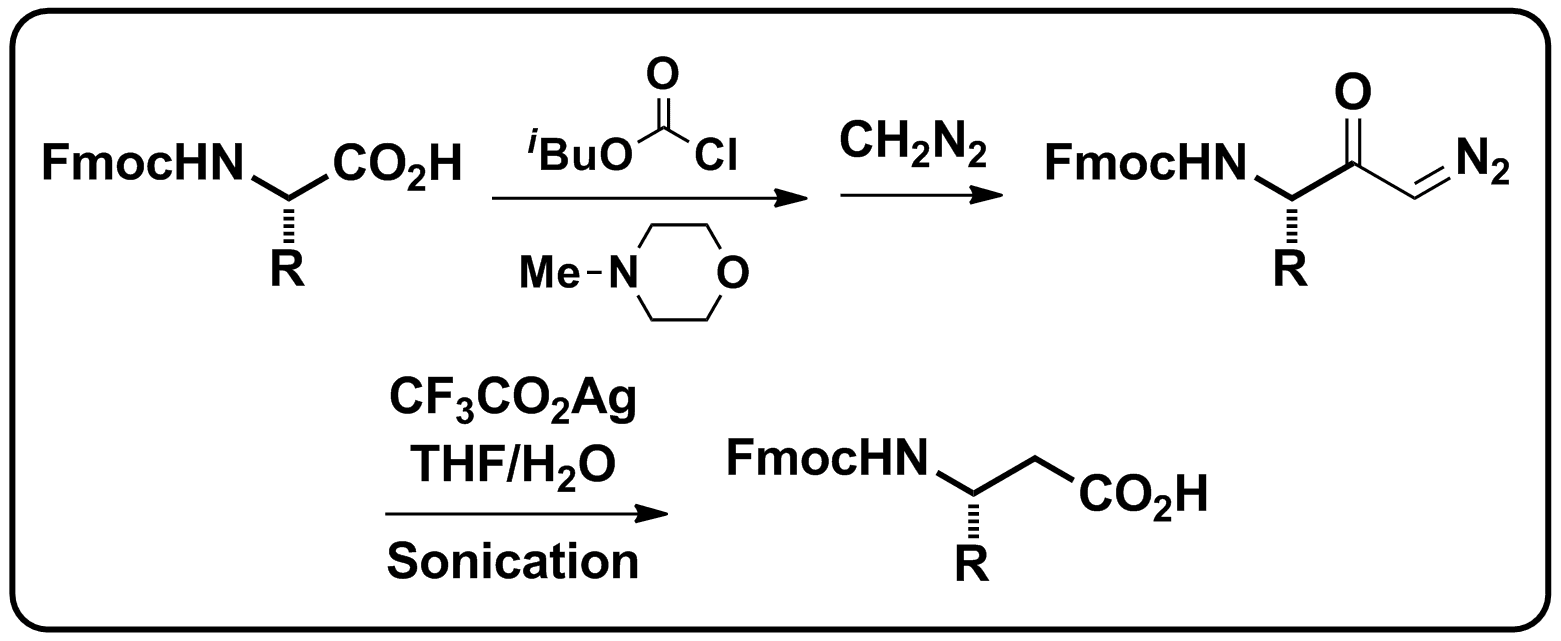
 For a change I had to leave the office, put on a lab coat and go to the lab to weigh out some compound. I had a nice fluffy freeze dried substance that had to be transferred from one vial to another. I was faced with usual static electricity problem. The easy solution is to take your plastic gloves off and hope that your compound doesn't fly around the hood when you try to transfer it. However, as often as not this will not do the trick. For the same reason the antistatic gun has been developed (see picture).
For a change I had to leave the office, put on a lab coat and go to the lab to weigh out some compound. I had a nice fluffy freeze dried substance that had to be transferred from one vial to another. I was faced with usual static electricity problem. The easy solution is to take your plastic gloves off and hope that your compound doesn't fly around the hood when you try to transfer it. However, as often as not this will not do the trick. For the same reason the antistatic gun has been developed (see picture).
If you "shoot" your vials a couple of times this will help, sometimes, maybe. I find it a bit of a lottery whether this works even without gloves on. Which brings me to the point of this post, the Mettler Toledo™ U-shaped Ionizer Antistatic System or as I call it "The Antistatic Portal". It reminds me of the movie Stargate as it's standing there next to your balance humming with electricity (see picture below). Basically the idea is that you just move the stuff that is being weighed through the portal and voila the problem is solved without travelling to distant planets and fighting Egyptian Gods.

Pretty neat and the damn thing actually works with plastic gloves and all so I can recommend this addtion to your lab (if you can afford it). D!

































 orcid.org/0000-0003-3926-7047
orcid.org/0000-0003-3926-7047

